Mild Cerebral Ventriculomegaly on Prenatal Ultrasound – Next Steps and Clinical Implications
CLINICAL ACTIONS:
According to SMFM recommendations, mild cerebral ventriculomegaly is identified on a prenatal ultrasound report when the atrium measures between 10-12 mm. Next steps include
Detailed anatomical study
Refer for genetic counseling
Consider diagnostic testing for aneuploidy
ACOG/SMFM guidance recommends offering invasive testing using microarray in the setting of fetal structural anomalies seen on prenatal ultrasound
Consider maternal toxoplasmosis and cytomegalovirus (CMV) testing regardless of whether the patient is aware of previous exposure
Consider fetal MRI for detection of associated cortical anomalies or signs of cerebral infection
May be of less value if a patient has already had an ultrasound examination by a professional with expertise in fetal brain imaging on ultrasound
Deliver based on standard clinical indications
If no abnormal findings detected on thorough examination, prognosis is favorable and the newborn is expected to be normal
Repeat ultrasound in the 3rd trimester, (30-34 weeks gestation)
Prognosis is better if measurement remains stable or improves/resolves
Serial ultrasounds unlikely to be helpful
Antepartum fetal testing not likely to be beneficial
Antiplatelet antibodies if intracranial hemorrhage also present
SYNOPSIS:
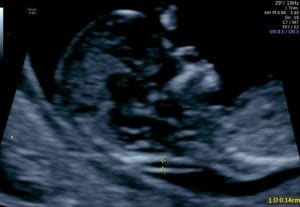
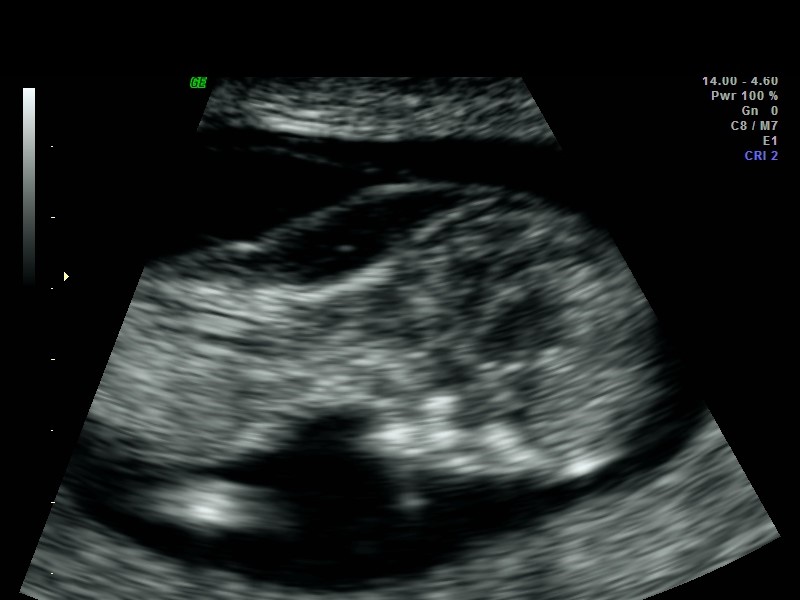
Mild/moderate ventriculomegaly may be seen in approximately 1% of fetuses. While mild cerebral ventriculomegaly may resolve or be of no consequence, clinical work-up is required to determine whether there is any physical obstruction, decreased cerebral volume or conditions that may alter production, reabsorption or cerebral spinal fluid (CSF) flow within the cerebral ventricles. Measurements of the lateral cerebral ventricles are obtained from an axial plane at the level of the thalamic nuclei.
KEY POINTS:
‘Fetal cerebral ventriculomegaly’ is present if the atrial diameter is ≥10mm and the SMFM further refines this definition as follows
Mild: 10-12 mm
Moderate: 13-15 mm
Severe: >15 mm
Ventriculomegaly is found in 0.15%-0.7% of chromosomally normal/euploid fetuses
When isolated, the incidence of an abnormal karyotype is 3.8% although some centers report higher rates
If patient opts for invasive testing, microarray is critical as copy number variants (CNVs) have been found in 8.3% of isolated cases following normal karyotype and were highly correlated with neurodevelopmental disorders
Ventriculomegaly is also associated with genetic syndromes, brain malformations, feto-neonatal alloimmune thrombocytopenia and congenital infections
Often, no cause will be found
Postnatal follow-up by pediatric neurologists and specialists should be considered
While children with a prenatal diagnosis of mild ventriculomegaly will overall have a very good prognosis, a few may still have abnormal neurodevelopment, dependent on associated anomalies and etiology
When only one ventricle is mildly enlarged and there are no other findings, chromosomal work up is more likely to be normal
There is still a risk of congenital infections (8.2%) and additional brain abnormalities (5% prenatal and 6.4% postnatal), as well as a risk of neurodevelopmental delay of 5.9%
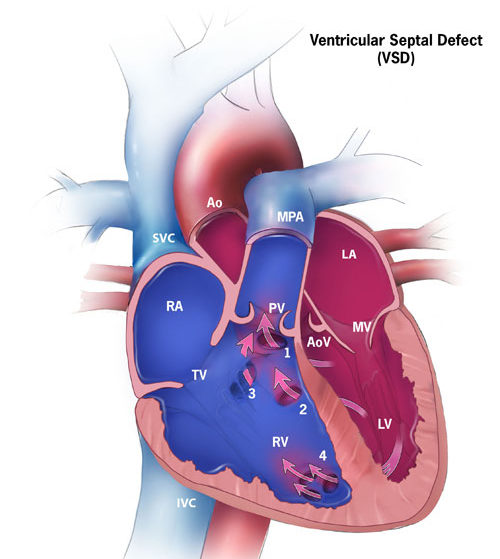
Ventricular Septal Defect – Implications for the Fetus
WHAT IS IT?
The left and right ventricles are connected through a “hole” in the wall/septum between the two chambers
Most common cause of all congenital heart defects (CHDs), comprising 50% of all CHDs
Estimated to occur in approximately 42/10,000 babies born
Isolated VSD accounts for 20% of all birth defects
Ventricular Septal Defects (VSDs) can be a single defect or multiple involving various components of the ventricular septum
The ventricular septum is made up of two major morphological components, muscular and membranous, and these components are further divided depending on location relative to other heart structures
KEY POINTS:
Cause of VSD is often unknown, but may be associated with common genetic issues such as Down syndrome (Trisomy 21) or 22q11.2 deletion syndrome, as well as more rare disorders
Can range from small isolated finding that will close on its own, to complex CHD requiring surgical repair and interdisciplinary team management
Consider genetic consultation and option for prenatal diagnostic testing to identify potential cause as may influence management
ACOG guidance recommends offering invasive testing using microarray in the setting of fetal structural anomalies seen on prenatal ultrasound
Further expert fetal imaging including fetal echocardiography is valuable to determine whether finding is isolated, as well as to determine prognosis and perinatal management plan
Image credit: Centers for Disease Control and Prevention, National Center on Birth Defects and Developmental Disabilities
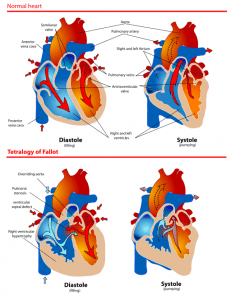
Tetralogy of Fallot- A Critical Congenital Heart Defect
WHAT IS IT?
Tetralogy of Fallot (TOF) is a congenital heart defect, that occurs in approximately 1 in every 2,500 live births and accounts for 7-10% of all congenital heart defects
TOF is comprised of 4 major abnormalities:
Pulmonary Infundibular Stenosis: narrowing of right ventricular outflow track
Overriding aorta: the aortic valve is connected to both ventricles
Ventricular Septal Defect: the left and right ventricles are connected through a “hole”
Right Ventricular Hypertrophy: the right ventricular wall muscle is thicker than usual
Cause is usually unknown, but may be associated with genetic issues
Consider 22q11.2 deletion syndrome and Down syndrome
ACOG guidance recommends offering invasive testing using microarray in the setting of fetal structural anomalies seen on prenatal ultrasound
Categorized as a ‘critical congenital heart defect’, which means a likelihood of surgery and intensive management and procedures during the first year of life
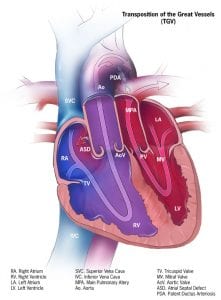
In a normal heart, the pulmonary artery carries deoxygenated blood to the lungs. Oxygenated blood returns to the left side of the heart and the aorta then pumps the oxygenated blood to the rest of the body. In Transposition of the Great Arteries (TGA), the pulmonary artery and aorta have changed places (i.e., they are transposed). Therefore:
The pulmonary artery, which usually arises from the right side of the heart and carries deoxygenated blood to the lungs, will now connect to the left side of the heart and send oxygenated blood back to the lungs
The aorta, which usually arises from the left (oxygenated) side of the heart, is now exiting the right side and therefore will carry deoxygenated blood to the rest of the body, bypassing the lungs
5 out of 10,000 babies are born with TGA
TGA is referred to as a ‘cyanotic’ (lacking oxygen) defect leading to babies with bluish discoloration and shortness of breath, with symptoms dependent on whether there is any ability for the deoxygenated and oxygenated blood to mix and be delivered to the rest of the body
Surgery is often necessary shortly after birth, especially in the case of complete TGA (also known as d-TGA referring to ‘dextroposition’) which is considered a ‘critical congenital heart defect’
Image credit: Centers for Disease Control and Prevention, National Center on Birth Defects and Developmental Disabilities
KEY POINTS:
While TGA can be diagnosed prenatally on ultrasound, it may not always be detected
In the majority of cases, a cause is not readily apparent
In some cases, TGA can be associated with genetic abnormalities and therefore, if a prenatal diagnosis is made or suspected, referral for genetic counseling is recommended, in addition to high risk obstetrical services, neonatology and pediatric cardiology
TGA is sometimes referred to as Transposition of the Great Vessels (TGV)
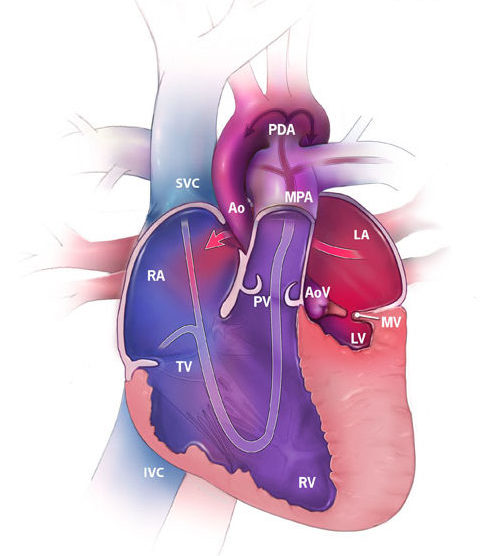
Hypoplastic left heart syndrome (HLHS) occurs when the left side of the heart fails to develop normally. As a result:
The left ventricle does not develop and is too small
Both the mitral valve and the aortic valve may not develop appropriately or are too small to function adequately
The ascending aorta is not well developed
A connection between the atria (artrial septal defect) or patent ductus arteriosus (small vessel that connects the aorta to the pulmonary artery) may be critical to keeping a newborn alive following delivery so that blood can circulate to the rest of the baby’s body
1 out of every 4,344 babies born in the U.S. have HLHS
Image credit: Centers for Disease Control and Prevention, National Center on Birth Defects and Developmental Disabilities
KEY POINTS:
In most cases, a cause is not found, however some genetic syndromes may be associated with HLHS, especially in the setting of other fetal structural anomalies
HLHS may be diagnosed prenatally on ultrasound
Infants may require multi-step operative procedures after birth, which are not curative, but rather have the goal of restoring function
Complex heart findings may ultimately require heart transplant for survival
Despite surgical interventions, HLHS is still associated with high mortality rates
While hospital survival has improved over the past decades, due to decreased cardiac output, post-operative morbidity remains significant and includes
Arrhythmias | Bleeding | Infection | Organ dysfunction (renal/hepatic)
Neurologic effects including seizures and neurodevelopmental delay
HLHS is considered a critical congenital heart defect and as such, a multidisciplinary approach is required if prenatal diagnosis of HLHS is made – including high risk obstetrics, neonatology, genetics and pediatric cardiology team
Duodenal Atresia – When the “Double Bubble” is Observed on Fetal Ultrasound
WHAT IS IT?
Duodenal atresia results from failure of recanalization of the duodenum after the 7th week of gestation due to an ischemic event or genetic factors.
The classic finding on ultrasound is the “double bubble sign” which is due to dilated proximal duodenum and stomach associated with lack of bowel gas in the distal intestine.
The incidence of duodenal atresia is 1/5000 – 1/10,000 live births
Polyhydramnios is present in 80% of cases of duodenal atresia
Trisomy 21 / Down syndrome is present in 25-40% of cases
Duodenal atresia can be associated with other congenital anomalies including:
VATER (vertebral defects, anal anomalies, esophageal atresia and renal anomalies)
Malrotation
Annular pancreas
Biliary tract anomalies
Cardiac anomalies
Mandibulofacial anomalies
KEY POINTS:
When detected, refer for
Fetal echocardiogram
Genetics consultation
ACOG guidance recommends offering invasive testing using microarray in the setting of fetal structural anomalies seen on prenatal ultrasound
Pediatric surgery consultation
Long term prognosis of isolated duodenal atresia is good, with 90% survival after surgery
Want to hear about the latest clinical summaries via ObG Insider?
Cystic Hygroma: Definition, Genetics and Prognosis
WHAT IS IT?
Cystic hygroma is a large single or multilocular fluid-filled cavity located in the nuchal region, behind and around the fetal neck, which can extend the length of the fetus and can be seen on fetal ultrasound. Cystic hygroma differs from nuchal translucency (NT). NT is a fluid-filled space normally seen behind the fetal neck on ultrasound performed in the first trimester. Unlike cystic hygroma, NT is a normal structure where size measurement is used as a risk factor for genetic disorders. A thick NT measurement is associated with aneuploidy and other structural anomalies.
CLINICAL ACTIONS:
Refer for high risk OB consultation and genetic counseling, and consider prenatal diagnostic testing
ACOG guidance recommends offering invasive testing using microarray in the setting of fetal structural anomalies seen on prenatal ultrasound
Detailed anatomic ultrasound
Fetal echocardiogram
SYNOPSIS:
A cystic hygroma is an anomaly of the vascular-lymphatic system formation. Failed venous-to-lymphatic connections lead to distended fluid-filled spaces and visualization of septations which may extend along the length of the fetal axis. A cystic hygroma can be seen as early as an 11-week ultrasound. Later in pregnancy, cysts may be seen on either side of the fetal neck, with a nuchal ligament running between the two structures. Aneuploidy is present in 50% of cases. There is a high rate of intrauterine fetal demise especially if hydrops is seen. However, 10 to 20% may resolve in utero in euploid fetuses.
KEY POINTS:
50% likelihood of aneuploidy
There is evidence that cystic hygroma detected early in the first trimester (< 45 mm CRL) may have lower rates of chromosomal abnormalities that those identified later in the first trimester (43% vs 73%)
Most commonly Trisomy 21 / Down Syndrome, 45X and Trisomy 18
<20% result in a healthy live-born infant at term
Not to be confused with nuchal translucency (NT)
Fluid filled space normally seen behind the fetal neck on first trimester ultrasound
NT measurement is a key component of fetal aneuploidy screening
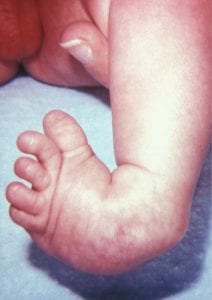
Clubfoot, congenital talipes equinovarus, is one of the most common orthopedic conditions diagnosed on prenatal ultrasound with an incidence of 1-3 per 1000 at birth.
WHAT IS IT?
In a fetus with clubfoot, the long axis of the foot (the sole) and the tibia can be seen in the same plane on ultrasound
False positive rate of clubfoot diagnosed by ultrasound may be as high as 10-19%
90% of clubfoot diagnoses are made in the first and second trimester
Detectable on ultrasound as early as 9 weeks gestation
Male predominance (M:F, 2:1)
Anatomy & Physiology, Connexions Web Site http://cnx.org/content/col11496/1.6/, Jun 19, 2013
KEY POINTS:
Underlying Causes
Genetic
Mendelian: Part of a genetic syndrome, especially a neuromuscular condition, such as arthrogryposis or congenital myotonic dystrophy
May be inherited as an autosomal dominant condition
Can occur in isolation
Chromosomal abnormality such as trisomy 18
Multifactorial birth defects such as neural tube defects that negatively impact fetal movement
Factors External to the Fetus
Environmental factors that restrict fetal movement such as
oligohydramnios
twinning
amniotic band syndrome
Bilateral vs. Unilateral
Bilateral vs. unilateral diagnosed prenatally in an approximately 1:1 ratio
Bilateral more likely to be confirmed at birth compared to unilateral (87.9% vs. 65.9%)
Bilateral is not more likely than unilateral to be associated with additional anomalies at birth
Overall in singletons, 11% with isolated club foot on prenatal ultrasound had additional findings at birth
Risk of Aneuploidy:
Risk of aneuploidy is increased in complex club foot (additional anomalies present)
Complex: approximately 30%
Isolated: Between 1.7% – 3.6% (literature suggests may be associated with sex chromosome aneuploidy)
ACOG guidance recommends offering
prenatal aneuploidy screening or diagnostic testing (amniocentesis or CVS) for all pregnant women regardless of age
Invasive testing using microarray in the setting of fetal structural anomalies seen on prenatal ultrasound
If suspected or diagnosed, refer for genetic counseling, MFM and pediatric orthopedic consultation for further discussion to determine possible etiologies, clubfoot correction, surgical and nonsurgical options
Cleft Lip and Palate – Considerations and Discussion Points
SUMMARY:
Orofacial clefts, which include cleft lip, cleft palate or combined cleft lip and palate are the most common congenital facial malformation.
Prevalence of orofacial clefts is approximately 1/700
Cleft lip with cleft palate (most common): 50% of cases
Isolated cleft lip: 25%
Isolated cleft palate: 25%
Majority are paramedian: 64% unilateral | 34% bilateral
More common in Asians American groups
Less common in African Americans
Facial clefts result from defective fusion of the facial processes
Association of facial cleft with other anomalies
13% of fetuses will have associated anomalies | Higher rate with bilateral and highest rate with median
Inheritance pattern
Isolated orofacial cleft typically multifactorial
Facial cleft may be associated with chromosomal and single gene disorders
CLINICAL ACTIONS:
Prenatal Detection
Detection rate of isolated cleft lip (with or without cleft palate) on ultrasound is variable across studies from 10% to 90%
Detection of cleft lip (with or without cleft palate) may be approximately 80%
Cleft palate is harder to detect and may be missed even by skilled sonographers
If bilateral cleft is identified, there is a higher risk for additional anomalies compared to unilateral cleft (25% to 10%, respectively)
Detailed prenatal anatomic ultrasound examination is recommended upon diagnosis
Evaluate for other anomalies to determine management and prognosis
Fetal echocardiogram should be considered
ACOG and SMFM recommend offering diagnostic testing
Microarray technology should be used due to finding of fetal structural anomaly (facial cleft) on ultrasound
Single gene testing or exome sequencing should be discussed with a genetic counselor on an individual basis
MRI may be an adjunct to prenatal diagnosis of orofacial clefts if there is concern for additional anomalies or if ultrasound imaging is incomplete
Treatment
There is no prenatal treatment
Consider pediatric surgical referral upon diagnosis to discuss potential surgical repair and appropriate setting for delivery
Genetics
In most cases, there is no clear identifiable cause of cleft lip
However, facial clefts have been associated with genetic syndromes and chromosomal anomalies
Referral to genetic counseling should be offered
Multifactorial Inheritance
Combination of genes and environment
Approximately 3% to 5% chance of recurrence which is 20% to 35% fold increase over baseline
If no other relatives based on professional pedigree analysis, risk to siblings may be lower than 3%
Recurrence risk increases
If more than one child affected
With increasing severity
Risk to other family members
Second degree relatives (half-sibs, aunts/uncles and grandparents): Much lower than above
Third degree relatives (e.g., first cousins): Likely not above baseline risk
Single Gene Disorders
Autosomal dominant, autosomal recessive and X-linked dominant disorders have been associated with orofacial clefts and should be considered if other family members are affected
May be syndromic, i.e., appears in a recognized constellation of other clinical findings rather than as an isolated birth defect
Chromosomal Disorders (rare if no other anomalies)
22q11.2 deletion syndrome
Submucosal Cleft Palate
Common autosomal trisomies
Trisomy 13 (associated with midline clefting)
Trisomy 18
Environmental Risks
Smoking
Second-hand exposure may be a risk factor
Alcohol consumption: Evidence not consistent
Binge drinking may be higher risk
Medications
Antiepileptics such as phenytoin, valproic acid
Folate antagonists (MTX)
Maternal pre-pregnancy diabetes mellitus
Obesity
KEY POINTS:
Isolated paramedian orofacial clefts without an associated genetic condition have an excellent prognosis with surgical repair
A median (midline) facial cleft has strong association with other anomalies and aneuploidy (Trisomy 13)
Mode of delivery and timing of delivery should not be impacted unless other usual obstetric indications
Omphalocele, or exomphalos, is the protrusion of internal organs, which may include intestines, liver (when present=giant omphalocele) and other abdominal organs, through the ventral wall of the fetus or infant and covered by a membrane consisting of peritoneum, Wharton’s jelly, and amnion
Results from failure of involved organs to return to abdominal cavity from the gut tube during 6-10 weeks of development
Estimated to occur in approximately 1/5400 births
Chromosomal abnormalities may be as high as 50%
Increased risk of chromosomal abnormalities related to presence of other anatomic anomalies or increased nuchal translucency
Trisomy 18 is the most commonly identified aneuploidy
Often associated with cardiac and/or neural tube defects
Image credit: Centers for Disease Control and Prevention, National Center on Birth Defects and Developmental Disabilities
KEY POINTS:
Cause of omphalocele unknown in most cases. Associations demonstrated with maternal history of:
Smoking
Alcohol use in pregnancy
Obesity
Prenatal Screening and detection is available:
Maternal Serum AFP (MSAFP) is elevated
Amniotic fluid AFP (AFAFP) is elevated
Can be detected on prenatal ultrasound
If an omphalocele is identified on sonography, consider:
Fetal chromosomal analysis including microarray as recommended by ACOG
Fetal echocardiogram referral
Genetic counseling to help identify potential causes, recurrence risk and management plan
Note: physiologic omphalocele may be noted in the first trimester but resolves by 12 weeks gestation
Liver should never be seen outside the fetal abdomen and this finding allows the diagnosis at any time in gestation
Refer to pediatric surgical specialist to discuss possible surgical correction options, outcomes and long term implications
Develop labor and delivery plan for those who will continue the pregnancy given high risk nature of pregnancy, and coordinate with NICU and pediatric team as needed
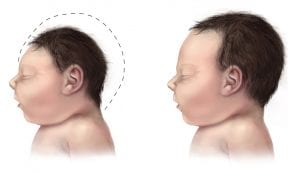
Microcephaly means “small head”, and can be used interchangeably in some instances with microencephaly, “small brain”
Consider abnormal development (primary microcephaly) if detected prior to 32 weeks gestation, as opposed to a degenerative condition in which normal head size becomes smaller (secondary microcephaly)
SYNOPSIS:
Microcephaly has an incidence of 2 to 12 in 10,000 births in the USA and can be diagnosed prenatally via ultrasound (in second or early third trimester) or postnatally via measurement of head circumference (HC). Microcephaly has been linked to developmental delay, seizures, as well as feeding, vision and hearing problems. Prognosis depends on the severity of the microcephaly and whether it is associated with other anomalies.
KEY POINTS:
Definitions
SMFM – Fetal Microcephaly
HC ≥ 3 SD below the mean for gestational age is the recommended definition
HC ≥ 5 SD below the mean is a certain diagnosis
CDC – Postnatal Congenital Microcephaly
Definite Congenital Microcephaly
HC at birth < 3rd percentile for gestational age and sex
If HC not available at birth, HC < 3rd percentile for age and sex within the first 2 weeks of life
For Stillbirths and elective terminations, CDC definition based on HC at delivery < 3rd percentile for gestational age and sex
Possible Congenital Microcephaly
For Live Births: If earlier HC is not available, HC < 3rd percentile for age and sex beyond 6 weeks of life
For All Pregnancy Outcomes: Microcephaly diagnosed or suspected on prenatal ultrasound in the absence of available postnatal HC measurements
American Academy of Neurology and Child Neurology Society
At birth
HC >2 SD below the mean for age and gender
Severe would be considered >3 SD below the mean
Management
If fetal HC by ultrasound is > 2 SD below the mean for gestational age, a careful evaluation of the fetal intracranial anatomy is indicated
If the intracranial anatomy is normal, consider follow-up in 3-4 weeks
When evaluating the finding of fetal microcephaly, assess for the following
Alcohol consumption, exposure to certain medications and/or smoking during pregnancy
Genetic conditions, including aneuploidy or single gene disorders
Many ultrasound packages report HC percentiles and not standard deviations (SD) and often the lowest reported HC measurement is <5th percentile
In such cases, SMFM suggests using head circumference (in millimeters) to determine SD below the mean
SMFM provides a table (see ‘Learn More – Primary Sources’ below) to determine the number of SD below the mean as a function of gestational age
Role of MRI: The SOGC Guideline states (III-A evidence)
Once fetal microcephaly is identified by ultrasound, fetal magnetic resonance imaging, when available and if potential findings are likely to alter pregnancy management, may be considered.
Fetal magnetic resonance imaging images should be reviewed by a radiologist with expertise in fetal magnetic resonance imaging.
Consider referral to a Fetal Maternal Medicine Specialist
Consider referral to Genetic Counseling, especially if microcephaly is associated with other findings
Consider referral to Pediatric Neurologist for review of short and long term potential outcomes for the newborn and child with microcephaly
Image credit: Centers for Disease Control and Prevention, National Center on Birth Defects and Developmental Disabilities
OBG Project CME requires a modern web browser (Internet Explorer 10+, Mozilla Firefox, Apple Safari, Google Chrome, Microsoft Edge). Certain educational activities may require additional software to view multimedia, presentation, or printable versions of their content. These activities will be marked as such and will provide links to the required software. That software may be: Adobe Flash, Apple QuickTime, Adobe Acrobat, Microsoft PowerPoint, Windows Media Player, or Real Networks Real One Player.
Disclosure of Unlabeled Use
This educational activity may contain discussion of published and/or investigational uses of agents that are not indicated by the FDA. The planners of this activity do not recommend the use of any agent outside of the labeled indications.
The opinions expressed in the educational activity are those of the faculty and do not necessarily represent the views of the planners. Please refer to the official prescribing information for each product for discussion of approved indications, contraindications, and warnings.
Disclaimer
Participants have an implied responsibility to use the newly acquired information to enhance patient outcomes and their own professional development. The information
presented in this activity is not meant to serve as a guideline for patient management. Any procedures, medications, or other courses of diagnosis or treatment discussed or suggested in this activity should not be used by clinicians without evaluation of their patient’s conditions and possible contraindications and/or dangers in use, review of any applicable manufacturer’s product information, and comparison with recommendations of other authorities.
Jointly provided by
NOT ENOUGH CME HOURS
It appears you don't have enough CME Hours to take this Post-Test. We no longer offer Hours.
Leaving ObG Website
You are now leaving the ObG website and on your way to PRIORITY at UCSF, an independent website. Therefore, we are not responsible for the content or availability of this site